Introduction
PhiSiGns consists of two critical, interlinked processes:
(1) Identification of signature genes conserved amongst a group of selected phages
(2) Design of PCR primers for the amplification of these signature genes
Web version
The convenient and user-friendly PhiSiGns web interface allows biologists to perform a dynamic search against selected phage genomes of interest, identify signature genes, generate nucleotide sequence alignments, and design primers for PCR amplification. The web version does not require additional input or third party programs.
Requirements
- Browser with JavaScript support
- Direct internet connection
To identify signature genes amongst phage genomes on PhiSiGns web version, please follow these steps:

- Click on options under "Limit display" to limit the display of available phage genomes
- Select phage genomes of interest from the displayed list of available phage genomes
- Add the selected phage genomes for signature gene identification and primer design
- Select the BLAST E-value cut-off for similarity searches
- Select the BLAST coverage cut-off for similarity searches
- Click on "Get Signature Genes" to get the list of identified signature genes (SiGs)
List of identified signature genes
Table of identified SiGs in the selected set of phage genomes

- Select desired SiG
- Click on "Continue" to design PCR primer pairs on a selected SiG
- Click on "Download short SiG table" to download the compact table
- Click on "Download detailed SiG table" to download the detailed table
- Click on "Show Genes" to display list of genes within a selected SiG
- Click on "Select Different Genomes" to revert to the previous step and change the selection of phage genomes
To design PCR primer pairs on the selected signature gene (SiG), please follow these steps:

- Adjust minimum and maximum primer parameter values [optional]
- Select or deselect genes within the selected SiG for alignment and primer design [optional]
- Download FASTA file for the selected SiG [optional]
- Upload your own nucleotide sequence alignment for the selected signature gene [optional]
- Click on "Show ClustalW alignment for selected genes" to view program generated alignment [optional]
- Click on "Design Primers for Selected SiG Genes" to get the list of potential primer pairs
List of potential primer pairs
Table of potential primer pairs for selected SiG

- Click on "Show Details" to display details for the selected primer pair [optional]
- Click on "Modify Primer Parameters or Genes Selection" to change the selection of primer parameters values or select/deselect genes within the selected SiG [optional]
- Click on "Download SiG Alignment" to download the nucleotide sequence alignment of the selected SiG [optional]
- Click on "Download Primer Pairs" to download the displayed primer pair table [optional]
- Click on "Download Primer Pairs Detail" to download the detailed primer pair table [optional]
Available options and parameters
E-Value
BLAST E-value represents the number of hits one can expect by chance when searching a database of a particular size. It is a statistical calculation based on the quality of alignment and the size of the database. The smaller the E-value, the more significant the similarity.
Coverage
BLAST coverage is the percentage of alignment length divided by the average query and subject sequence length. The larger the coverage, the more significant the similarity.
SiG Function
cd
Dominant functional role of the genes within a SiG
SiG Length
Mean nucleotide sequence length of the genes within a SiG
Primer Length
Number of bases in the primer.
GC content
The number of G's and C's in the primer as a percentage of the total bases.
Melting Temperature (Tm)
The temperature at which one half of the DNA duplex will dissociate and become single-stranded. Primer melting temperature is computed using three methods:
Basic Melting Temperature: The Basic Melting Temperature calculation is done using the equations from Marmur (1962), and Wallace et al., (1979) [1,2].
For primers less than 14 nucleotides:
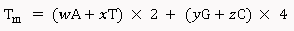
For primers longer than 13 nucleotides:
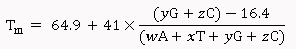
Where, w,x,y,z are the number of the bases A,T,G,C in the sequence, respectively.
Salt-Adjusted Melting Temperature: The Salt-Adjusted Temperature calculation is done using the equations from Nakano et al., (1999), and Howley et al., (1979) [3,4].
For primers less than 14 nucleotides:
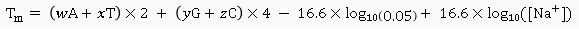
For primers longer than 13 nucleotides:
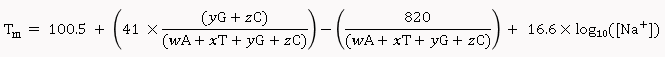
Where, w,x,y,z are the number of the bases A,T,G,C in the sequence, respectively.
Nearest-Neighbor Melting Temperature: The Nearest-Neighbor calculations are done essentially as described by SantaLucia (1998) [5]. The melting temperature is given by the equation:
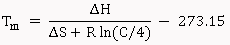
Where, ΔH (kcal/mol) is the enthalpy of nearest neighbor interactions, ΔS (cal/K-mol)is the entropy of nearest neighbor interactions, C is the molar concentration of the primer, and R is the gas constant (1.987 cal/K-mol). Both ΔH and ΔS are computed by summing all nearest neighbor base pair interactions. The nearest neighbor parameter values for ΔH and ΔS for DNA/DNA duplexes were obtained from SantaLucia (1998) [5]. An additional salt correction is added as,
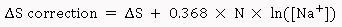
Where, N is primer length-1 and Na+ is the salt concentration (mM).
The calculations above assumes 200 nM [primer], and 50 mM [Na+]. The calculations also assume that the primers are not symmetric.
3'GC Clamp
The presence of G's or C's within the last five bases from the 3' end of a primer. Primers with more than 3 G's or C's in the last bases at the 3' end of the primer will be ignored.
Minimum delta G
The Gibbs free energy (ΔG kcal/mol) is the measure of the spontaneity of the reaction, representing the energy required to break the secondary structure. Larger negative values for ΔG indicate more self-priming and stable, undesirable hairpins. Minimum ΔG of a forward and a reverse primer is computed using the equation

Where, ΔH (kcal/mol) is the sum of nearest neighbor enthalpy changes. ΔS (cal/K-mol) is the sum of nearest neighbor entropy changes. T is the temperature in oC (37oC) and is converted to Kelvin by adding 273.15.
Maximum 3' stability
The maximum stability for the five 3' end bases of a forward and reverse primer. Primers with ΔG ≥ -9 kcal/mol for the five bases from the 3' end will be considered.
User-generated alignment
This option is to allow the users to refine and upload a SiG alignment using alignment tools of their choice. The alignment must be uploaded in 'CLUSTALW' format with '.aln' file extension. By default, PhiSiGns generates CLUSTALW sequence alignments with default parameters.
Complementarity
Primer pairs are tested for complementarity:
1) Dimers: A primer self-dimer is formed by intermolecular interactions between two of the same primers i.e., forward vs. forward or reverse vs. reverse. Cross-dimers are formed by intermolecular interactions between a forward and a reverse primer. Primer dimers with more than 4 consecutive base pairings are not passed and will not be considered as potential primer pairs.
2) Hairpins: Hairpins (with a minimum 3 base pair loop and 4 base pair stem length) are formed by intramolecular interaction within the primer. Primer hairpins with more than 4 consecutive base pairings are not passed and will not be considered as potential primer pairs.
Primer pairs that meet the above criteria for self-dimers, cross-dimers and hairpins are tagged as 'Pass'. Primer pairs that do not qualify the above criteria for all three tests are tagged as 'Fail'. Primer pairs that meet the criteria for one or two out of the three tests are tagged with 'Warning'.
Product size
The product length is the target length for PCR and is calculated as: (Start position of forward primer-End position of reverse primer) +1.
Degeneracy
Degeneracy of a primer sequence is computed by multiplying degeneracy of each contributing IUPAC nucleotide code. For example, a primer with the sequence GRBNA would correspond to a degeneracy of 1x2x3x4x1=24
| IUPAC nucleotide code | Degeneracy |
|---|---|
| A C G T | 1 |
| R Y S W K M | 2 |
| B D H V | 3 |
| N | 4 |
Tm Mismatch
The forward and reverse primer of a primer pair should have a temperature difference of 10oC or less. The Tm mismatch difference is an average computed over the three melting temperature calculations.
Standalone version
The standalone version is a perl script (phisigns.pl) and requires installation of Perl/BioPerl modules, and software packages for signature gene identification and PCR primer design. The README file has the entire prerequisite and information on the usage of the standalone version. The PhiSiGns standalone version is available at http://sourceforge.net/projects/phisigns/files/.
Resources
- Direct internet connection
Software
- BLASTALL (BLAST standalone package)
- CLUSTALW 1.8 (Multiple sequence alignment tool)
BioPerl modules
- Bio::Seq
- Bio::SeqIO
- Bio::SeqFeature::Generic
- Bio::DB::GenBank
- Bio::DB::Fasta
- Bio::Index::Fasta
- Bio::Tools::Run::StandAloneBlast
- Bio::Tools::Run::Alignment::Clustalw
- Bio::Align::Utilities qw(aa_to_dna_aln)
- Bio::AlignIO
Perl modules
- Getopt::Long
- File::Basename
- List::Util qw(max)
Files
- input.txt
| Option | Description | Default | Argument |
|---|---|---|---|
| -d | PhiSiGns working directory path | - | string |
| -m | run mode | 0 | int |
| -e | BLAST E-value cut-off | 10 | float |
| -c | BLAST coverage cut-off | 10 | float |
| -i | input file | - | string |
| -g | selected SiG_# for primer design | - | string |
| -a | user uploaded alignment file | - | string |
| -aln_only | Display default ClustalW alignment | - | none |
| -id | id of current job | - | string |
There are two modes to run PhiSiGns:
Mode 0
Identifies signature genes amongst the user-selected phage genomes as listed in the input file ('-i') saved under the PhiSiGns working directory. The input file must include the NCBI refseq number for the phage genomes to be compared for signature gene identification and primer design. Type the following command line to execute phisigns.pl in Mode = 0. BLAST E-value ('-e') and coverage ('-c') cut-off can be set by the user; if not specified a default value of 10 and 10 is used, respectively.
% phisigns.pl -d /home/user/phisigns/ -m 0 -e 0.1 -c 25 -i input.txt -id test
Once the above command is executed and finished running, data and result files are generated with respect to the job id specified ('-id') in the command line under the PhiSiGns working directory.
Mode 1
Design PCR primer pairs for a selected signature gene from the list of identified SiGs generated in Mode = 0. The primers are designed taking into consideration the minimum and maximum values specified for the primer parameters. Users can modify the values of these parameters to their preference in the primer parameter file (pparams.txt).
| Option | Description | Default | Range |
|---|---|---|---|
| minl | min primer length | 16 | 10-28 |
| maxl | max primer length | 28 | 10-28 |
| mingc | min GC content | 30 | 20-80 |
| maxgc | min GC content | 30 | 20-80 |
| minbtm | min Basic Melting Temperature | 30 | 30-80 |
| maxbtm | max Basic Melting Temperature | 80 | 30-80 |
| minstm | min Salt Adjusted Temperature | 30 | 30-80 |
| maxstm | max Salt Adjusted Temperature | 80 | 30-80 |
| minnnh | min Nearest Neighbor Temperature | 30 | 30-80 |
| maxnnh | max Nearest Neighbor Temperature | 80 | 30-80 |
| mindg | min delta G for a primer | -20 | -30-0 |
| minpl | min product length | 400 | 100-2500 |
| maxpl | max product length | 2000 | 100-2500 |
| dgn | primer degeneracy | 1000 | 1-1000 |
| gcclamp | 3'GC clamp | y | y,n |
| max3stb | maximum 3' stability | y | y,n |
| compl | Complementarity | y | y,n |
Type the following command line to execute phisigns.pl in Mode = 1 to only display program generated alignment for a selected signature gene:
% phisigns.pl -d /home/user/phisigns/ -m 1 -g SiG_5 -aln_only -id test
Type the following command line to execute phisigns.pl in Mode = 1 with the program generated alignment:
% phisigns.pl -d /home/user/phisigns/ -m 1 -g SiG_5 -id test
Users can also upload their own nucleotide sequence alignment (in CLUSTALW format only with '.aln' file extension) for the selected signature gene group to design PCR primer pairs. The user uploaded alignment file should be under the phisigns working directory. Type the following command line to execute phisigns.pl in Mode = 1 with a user-generated alignment:
% phisigns.pl -d /home/user/phisigns/ -m 1 -a test.aln -id test
Once the above command is executed and finished running, result files for the user uploaded alignment are generated with respect to the job id specified ('-id') in the command line under the phisigns working directory. PhiSiGns (phisigns.pl) in Mode = 1 will execute only after PhiSiGns is executed in Mode = 0. Unless the selected set of phage genomes is different, Mode = 0 need not be executed more than once. To design PCR primer pairs with a user-uploaded alignment, Mode = 1 can be executed without running Mode = 0 first.
Standalone vs. Web version
Both the standalone and web versions provide the same features, but differ in the following:
- The standalone version does not come with a phage genome database or precalculated BLASTP outputs. The user inputs the phage genomes to be compared for signature gene identification and primer design in an input txt file. The phage genomes are input as GenBank accession numbers only (see Standalone version - running mode). The GenBank files for the user-selected set of phage genomes are then obtained from NCBI and the gene annotations for the protein coding regions are imported accordingly. In contrast, the PhiSiGns web version consists of a database of 636 phages and 33 archaeal viruses (derived from the phage database on the PhAnToMe website, Feb 2011 [http://www.phantome.org/Downloads]), and pre-calculated BLASTP outputs for all these genomes (computed using PhiSiGns at an E-value cut-off of 10). The gene annotations are imported from the SEED [http://www.theseed.org/wiki/Home_of_the_SEED] and the proteins that lack annotation are extracted from GenBank. Therefore, with the standalone version, users have the ability to compare phage genomes that are not part of the web-based PhiSiGns phage database.
- The standalone version does not provide the option to select/deselect genes within the selected signature gene group for alignment and primer design. However, users can upload their own alignment containing only the sequences of interest.
References
- Marmur J, Doty P: Determination of base composition of deoxyribonucleic acid from its thermal denaturation temperature. J Mol Biol 1962, 5:109-118.
- Wallace RB, Shaffer J, Murphy RF, Bonner J, Hirose T, Itakura K: Hybridization of synthetic oligodeoxyribonucleotides to Phi X174 DNA - effect of single base pair mismatch. Nucleic Acids Res 1979, 6:3543-3557.
- Nakano S, Fujimoto M, Hara H, Sugimoto N: Nucleic acid duplex stability: influence of base composition on cation effects. Nucleic Acids Res 1999, 27:2957-2965.
- Howley PM, Israel MA, Law MF, Martin MA: Rapid method for detecting and mapping homology between heterologous DNAs - evaluation of polyomavirus genomes. J Biol Chem 1979, 254:4876-4883.
- SantaLucia J: A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci USA 1998, 95:1460-1465.